Regulatory triangles, open doors, and the cost of waiting for someone else to go first.
MO:RE spoke with Jeffrey Brown, Senior Scientist for Pharmaceuticals and Medical Devices at the PETA Science Consortium International, in December 2025.
Jeff Brown didn’t plan to spend seventeen years working on animal testing alternatives. Before joining PETA, he was a bench scientist in molecular biology and immunology, and increasingly disillusioned. “I had that internal view of watching how curious results, low statistical power, how all of that was massaged in grant applications.” The story of how he ended up in regulatory science starts, improbably, on a flight from DC to Los Angeles, where he sat next to the woman who’d built PETA US’s regulatory toxicology department from scratch. By the time they landed he had a job interview. That was 2007. By 2012, the organisation had founded the Science Consortium International and by their 10-year anniversary built a team of 25 scientists working across regulatory toxicology, chemical safety, and pharmaceutical development.
What surprised us most about our conversation wasn’t any single claim Jeff made. It was his framing of the entire landscape: a system where everyone agrees things need to change, and everyone is pointing at someone else to go first.
An awkward triangle
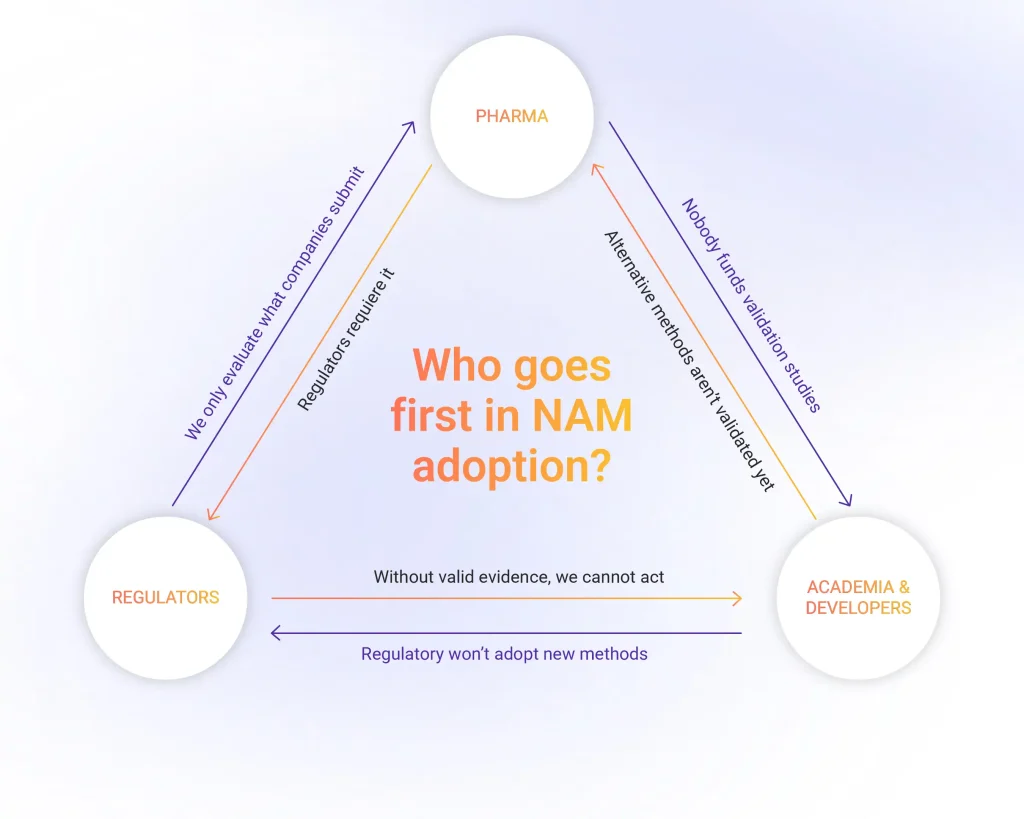
Jeff describes the regulatory landscape around animal testing alternatives as a triangle. On one side, government agencies: FDA, EMA, OECD, national regulators. On another, regulated companies: pharma, chemicals, consumer goods. On the third, everyone else: academic researchers, technology developers, advocacy organisations, the public.
“Without fail,” he says, “everyone points to another responsible party.”
Ask a pharma company why they still run animal studies and they’ll tell you the regulators require it. Ask the regulators and they’ll tell you they can only evaluate what companies submit, and companies keep submitting animal data. Ask the academic researchers developing alternatives and they’ll tell you nobody’s funding the validation studies that would make their methods regulatory-grade.
The striking thing about this isn’t that it’s a coordination failure. Those are everywhere. It’s that all three sides are, individually, telling the truth. The confusion, Jeff argues, is structural. These institutions are staffed by individual humans with varying expertise, willingness, and institutional memory. Key personnel retire and take decades of knowledge with them. Replacement hires may not share the same priorities, or may simply not know what their predecessors knew about how the system actually works.
“The confusion is such an essential part of this,” Jeff says, “that everyone needs to sometimes take a deep breath.”
The doors are open
Here’s the thing though: the formal regulatory barriers to replacing animal tests are, in many cases, lower than the operational ones.
In Europe, Directive 2010/63/EU established a legal requirement that animal procedures be replaced when scientifically satisfactory alternatives exist. That’s not a suggestion. It’s the law. It has been the law for over a decade.
In the US, the picture shifted more recently. The FDA Modernization Act 2.0, signed in December 2022, didn’t ban animal testing. What it did was replace “preclinical tests (including tests on animals)” with “nonclinical tests” throughout the statute, and then defined nonclinical tests to explicitly include cell-based assays, microphysiological systems, computer models, and animal tests (S.5002). It expanded the menu of acceptable evidence without removing anything from it.
“That didn’t necessarily change anything,” Jeff says. “It didn’t create any new rights or responsibilities for the FDA.”
But it opened doors. And by the time we sat down with Jeff, the FDA had already started walking through them. In April 2025, the agency announced a plan to phase out animal testing requirements for monoclonal antibodies, accompanied by a five-year roadmap to reduce animal studies from the norm to exceptions. Days before our conversation, they’d issued draft guidance for monospecific antibodies framing chronic primate toxicity studies as “generally unwarranted”, with provisions to eliminate them entirely in certain cases.
The assertion gap
So if the doors are open, why isn’t everyone walking through?
Jeff’s answer is blunt: most companies don’t know how to talk to regulators about alternatives, and most regulators can’t proactively tell companies what to do differently.
“Practice being very assertive about what you’re saying,” he advises. “You know this. You know it so much better than any regulatory reviewer will know.”
This cuts against the instinct most scientists bring to regulatory interactions. The academic training is to present data cautiously, hedge conclusions, invite critique. But regulatory submissions don’t work like peer review. You’re not proposing a hypothesis; you’re making an assertion about the safety or efficacy of a product, and the agency’s job is to evaluate whether your evidence supports that assertion. If you come in tentatively, you get tentative responses.
“You should meet at where your strong assertion is,” Jeff says.
There’s an economic dimension too, and not just the cost of submissions. “Every pharmaceutical company probably doesn’t speak about it very loudly,” Jeff says, “but internally they know very well; they have probably failed many drug candidates through preclinical testing that they don’t believe would have been harmful in humans.” The current system doesn’t just miss toxic drugs that reach the market with black-box warnings. It also kills good drugs that never get the chance. That’s an enormous back catalogue of substances waiting to be reassessed and a powerful incentive for companies that can build credible non-animal evidence packages.
The cost of engaging regulators compounds the problem. Taking a submission to the FDA is expensive: preparation, legal review, the submission itself, follow-up meetings. Companies can’t afford to run exploratory conversations at full regulatory submission cost. Jeff’s organisation helps bridge this gap through early, lower-cost pre-submission engagements with agencies. What he describes as finding “avenues to make sure that you understand what the competencies are at a regulator, where you may need to be prepared to be more of a teacher.”
That last phrase is worth sitting with: Where you may need to be more of a teacher. The assumption in most industry-regulator interactions is that the agency holds the expertise and the company is seeking approval. For novel methodologies like organoids, organ-on-chip, and computational models, the dynamic is inverted. The company developing the method often understands it better than any individual reviewer. The challenge isn’t getting permission. It’s communicating clearly enough that the reviewer can evaluate your assertions on their merits.
First movers
Jeff makes a distinction we found useful. Developing new approach methodologies from first principles is genuinely hard. The biology, the engineering, the validation science. But recognising where current methods fail? “That happens at a much lower level.”
The chemicals space is the clearest proof. There are hundreds of thousands of synthetic chemicals in circulation. Animal-based testing can accommodate perhaps 5,000 per year, a minute fraction of the possibilities in chemical space. “It’s easy to look at that math,” Jeff says, “and say, we’re never going to get there.” That arithmetic, not ethics, not politics, is what forced the chemicals sector to rebuild its testing infrastructure. The OECD now maintains a large and growing body of test guidelines that don’t use animals, covering endpoints from skin irritation to acute toxicity to mutagenicity. The Science Consortium tracks all validated non-animal methods accepted for regulatory use. And the list keeps getting longer.
The pharmaceutical space is grappling with a harder version of the same problem. Not just safety but efficacy, not just “is it harmful” but “does it work”. But the precedents are stacking up.
Novo Nordisk established a task force in the early 2000s to review animal use in their manufacturing quality control. Nobody required this. What they found was that many animal-based batch tests were redundant: holdovers from before cell-based assays could deliver equivalent signals. By 2012, they had eliminated animal testing from biological product batch control entirely, down from over 13,000 animals per year in the 1990s to zero. The key: not a single replacement method, but process improvements and cell-based assays that made the animal tests redundant.
Eli Lilly followed a parallel path on pyrogen testing. Starting in 2016, they began converting from the limulus amoebocyte lysate (LAL) test, which relies on horseshoe crab blood, to recombinant Factor C (rFC), a synthetic alternative. By 2018 they had their first regulatory approval for an rFC-tested product. Nobody mandated the switch. They chose it, proved it worked, and talked about it publicly.
And on January 1, 2026, just weeks after our conversation, the European Pharmacopoeia’s suppression of the rabbit pyrogen test (Chapter 2.6.8) took full effect. Approximately 400,000 rabbits were used annually worldwide for this single test. Now, that number will plummet. Not because of a scientific breakthrough, since the replacements have been validated and available for years, but because of the institutional commitment to stand behind the alternatives.
“A rare achievement,” Jeff says, “that a massive project in this space is welcomed, successful, and early.”
There’s a pattern across these examples: invest in better process, demonstrate reliability, earn regulatory endorsement, gain competitive advantage.
Our corner of the triangle
We asked Jeff this directly.
MO:RE is building the end-to-end automation infrastructure for organoid research. Not a single assay or a single animal test replacement, but the instrumentation, standardised culture, and process control, that sit underneath all of them. Our bet is that the transition Jeff had been describing won’t happen lab by lab, each team solving reproducibility on their own. It’ll happen when there’s a common infrastructure layer that makes organoid evidence trustworthy by default. Cloud computing didn’t just make servers cheaper but changed who could build software at all. Where does that sit in the triangle?
Jeff’s answer reframed how we think about our own work.
“I’ll even expand from automation to process,” he said. “The process controls, the consistency of manufacture has become much more important than the specific tests that are conducted.”
He pointed to vaccines as the clearest parallel. Vaccines are biological products, each batch could theoretically be different. The historical approach was to test each batch extensively, including with animal-based potency and safety assays. The modern approach is to control the manufacturing process so tightly that consistency is guaranteed by the process itself, not by endpoint testing. Strong process control plus many small automated checks replaces the need for heavy batch-level animal tests.
“It’s left to the engineers,” Jeff says, “and yet very strong production consistency is widely understood to be the path, an unavoidable path.”
That’s one leg of it; process consistency as evidence. But the conversation also surfaced a second leg, which connects back to the throughput arithmetic Jeff raised about chemicals. New approach methodologies solve the what: better models, more human-relevant biology, disease-specific endpoints. But they don’t automatically solve the how. If the testing backlog is measured in hundreds of thousands of compounds, and if the regulatory case for NAMs depends on demonstrating reproducibility across conditions, operators, and sites, then automation isn’t a convenience layer on top of the biology. It’s what makes the biology scalable enough to matter.
The biological case for organoids is being made by thousands of labs worldwide. What’s often missing is the process consistency that makes that case credible to regulators: the reproducibility, the audit trails, the statistical process control. If the bottleneck isn’t the biology but the credibility of the biology—if regulators need to trust that your organoid behaves the same way on Tuesday as it did on Friday, across sites, across operators—then process consistency isn’t a nice-to-have. It’s the regulatory case itself. We’re not only building convenience. We’re building the path to reliable evidence.
The long view
We asked Jeff what he’d change if he had a magic wand. His answer was immediate: “One key person at all times who was a decision maker.” Every struggle, in his experience, comes back to the absence of clear decision authority. The triangle pointing at itself in perpetuity.
The triangle doesn’t need a single decision maker, but rather people in each corner willing to move first and be assertive about why.
The regulatory landscape for animal testing alternatives is moving faster than at any point in the last four decades. The FDA has gone from opening doors to actively showing people where to walk. The European Pharmacopoeia just completed a landmark phase-out ahead of schedule. Companies that invested in process improvements a decade ago are reaping the advantages. And the tools to make non-animal methods reproducible are maturing rapidly.
Jeff’s advice to companies in this space is simple: don’t wait for someone else in the triangle to go first. Assert what you know. Show your work. And don’t keep it to yourself.
“There’s no benefit to keeping your struggles and your successes secret,” he says. “Talk about it.”
So we’re talking about it.